FragmentDB Plugin
FragmentDB is a powerful plugin tool for fragment fitting during refinement with Olex2. It extracts pre-constructed molecular fragments such as common solvents, anions, and cations from a database and fits them into an existing model. Users can add their own fragments as well. FragmentDB can greatly simplify dealing with, e.g., a disordered solvent or ion on a special position.
FragmentDB is part of Olex2 and located in the tools menu, so there is no download or installation necessary.
Literature
-
D. Kratzert, I. Krossing, J. Appl. Cryst. 2018, 51, 928-934. doi: 10.1107/S1600576718004508 download
-
D. Kratzert, J.J. Holstein, I. Krossing, J. Appl. Cryst. 2015, 48, 933-938. doi:10.1107/S1600576715005580 download
Fragment Chooser
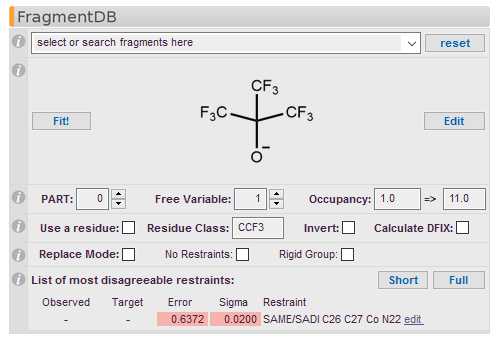
To start, choose a fragment from the drop-down menu. Clicking on a name in the list selects the fragment to be fitted into the model and displays its chemical structure in this panel.
To search for a particular fragment in the database, type in part of its name and hit Enter. For example, a search for 'CF3' finds all fragments containing a trifluoromethyl group, as well as structurally similar fragments. Select the desired fragment from the shortened drop-down menu containing the search results.
Reset
The reset button discards the selected fragment and restores the full fragment list after a search.
User Defined Database
The main database is write-protected, but it is possible to add new fragments to the
collection or change existing fragments. They will be stored in a separate database
in Datadir()/db/user-fragment-database.sqlite.
Type 'shell DataDir()/db' to
open this folder. The user fragment database will never be overwritten by any Olex2
plugin updates!
Fragments created or changed by the user will get a *user* tag at the end of their
names. Any database editor capable of handling SQLite files can be used to modify
the user fragment database.
Picture
Displays the chemical structure of the fragment. Click on the structure to display a magnified version. User-defined fragments will only display either an auto-generated structure or a user-defined drawing that can be selected in the fragment editor.
Fit!
The Fit! button starts the process of fitting the chosen fragment into the structure. The fragment appears on the screen in Mode Fit, and can be moved or rotated as desired. Press Escape to exit the mode.
Edit
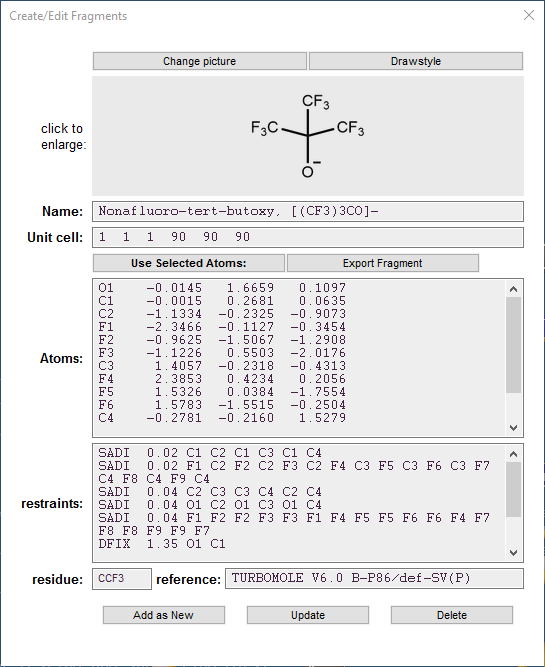
Clicking this button will open an editor in which the currently selected fragment can be modified. The editor can also be used to create and enter new fragments in the database.
Editor Options
Change Picture
Use a different picture for the chemical structure of the fragment. Pictures should have a resolution of at least 600 dpi.
Drawstyle
Saves a ChemDraw style file for drawing fragment structures with the same appearance as the predefined ones.
Name
Name of the fragment.
Unit cell
Cell parameters of the structure from which the fragment was obtained. Use 1 1 1 90 90 90 for rectangular Cartesian coordinates. The cell constants are used to calculate Cartesian coordinates for atoms of a fragment being added to the database.
Use selected atoms
Uses the selected atoms for the Atoms field and creates a picture of the fragment's chemical structure.
Atoms
Insert atoms here manually, as, for example: O2 8 1.3984 -0.3778 0.4922. It is possible to copy atom symbols and coordinates directly from SHELX files or copy coordinates generated by the free and open-source molecule editor Avogadro available at http://avogadro.cc/. Nevertheless, the simplest and least error-prone method is to use the 'Use selected atoms' button.
Restraints
Type any SHELX-compatible restraint(s) for the fragment in this field, except SAME. The length of a line is unlimited. Thus, for example, a CF3 group has the following restraints:
SADI C1 F1 C1 F2 C1 F3
SADI 0.03 F1 F2 F2 F3 F3 F1
Residue
Defines the residue class of the fragment. This is a brief code of up to four alphanumeric characters identifying the fragment; the first has to be a letter. If the fragment is incorporated into a model using a RESI command, this class will be the default name of the residue.
Properties
Define PARTs, free variables (FVAR) and Occupancy for the fragment here. The value of the free variable and occupation factor combination as it appears in the SHELX instruction file is automatically generated from the input values and displayed in the last field of this line. For example, the free variable index 31.0 in SHELX notation specifies free variable (FVAR) 3 and a multiplicative factor of 1.0, for a site occupancy equal to FVAR 3. On the other hand, -31.0 also specifies free variable 3, with a multiplicative factor of -1.0, i.e, the site occupancy will be (1-FVAR 3), to ensure that the total site occupancy always sums to 1.
Parameters of the fitted fragment such as PART number, FVAR, occupancy, and residue number (if any) are displayed with other atomic information in the structure on the screen when any parameter of PART, FVAR, occupancy or residue is changed. The residue number is separated from the atom label with an underscore, e.g., C3A_1 means atom C3A in residue 1.
Use a residue
Enable fitting the fragment as a residue, using a RESI command in the .ins file, by ticking this box. The Residue Class is the name of the residue. The default name provided by the database generally suffices, but any desired name for the residue can be entered in the Residue Class box. The name can be up to four characters long and must start with a letter.
Invert
Inverts the coordinates of the fragment. If this box is ticked, the geometry of the fragment will be inverted before being used in fitting.
Calculate DFIX
Fragments already in the database normally include predefined restraints such as SADI, FLAT, RIGU, etc. Ticking the Calculate DFIX box calculates DFIX/DANG restraints from the geometry of the fragment and replaces all predefined restraints with just the calculated DFIX/DANG restraints. The ADPs in the resulting fragment will not be restrained in any way, so the thermal ellipsoids of atoms in the fragment may appear unusual during refinement.
Replace Mode
If enabled, all atoms in PART 0 lying within 1.22 Å of the atoms of the fitted fragment will be deleted. This is particularly useful to replace atoms of a disordered structure just solved using SHELXT. It will not delete atoms in PARTs other than those in PART 0.
No Restraints
No restraints will be applied to the fitted fragment. This may lead to problems in refinement.
Rigid group
Keeps all atomic coordinates of the fragment fixed during refinement.
Revert Fit
Goes back in Olex2 refinement history to the step just before the last fragment fit.
Results
This table contains a list of the most disagreeable restraints (if any). Clicking on one of the lines in the list opens an editor window in which to adjust the parameters of that restraint. Short only shows the most disagreeable restraints, while Full shows the complete list of all restraints.
The error of a parameter should generally not exceed 3σ. A larger error means that the value of the parameter is not justified by the experimental observations. The reasons for this error might be, for example, a distorted molecular geometry such as a bend in an aromatic ring or systematic errors in the data. Disagreeable restraints will be highlighted in yellow if the error is above 2.5σ, and in red if the error is above 3.5σ.